|
Developed by 
|
Supported by 
|
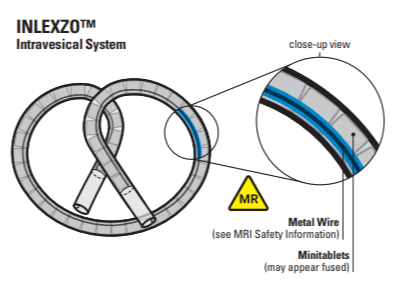
Gemcitabine Intravesical LAI
Developer(s)

|
Johnson & Johnson Originator
https://www.jnj.com/innovativemedicine/
United States of America Janssen Biotech, Inc. is a leading biotechnology subsidiary of Johnson & Johnson (J&J) that specializes in developing, manufacturing, and commercializing therapeutic products to treat serious medical conditions. As of 2026, the company is part of the Johnson & Johnson Innovative Medicine division, following a global rebranding initiative to unite J&J's pharmaceutical segments under a single brand |
Drug structure
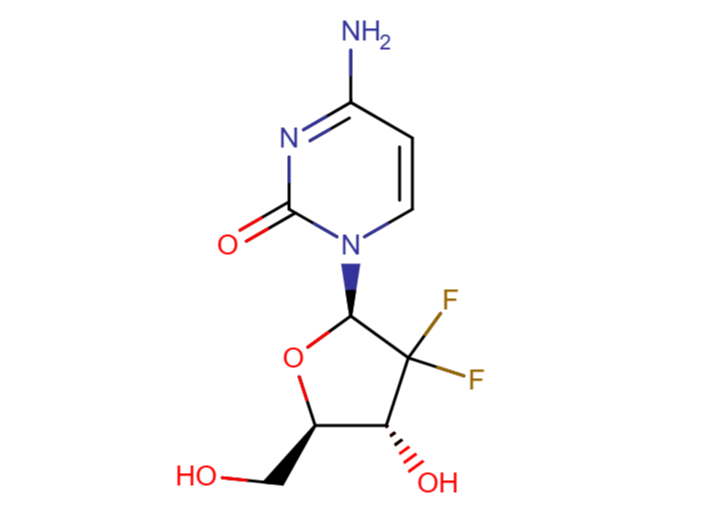
Structure of Gemcitabine
https://go.drugbank.com/drugs/DB00441
Gemcitabine Intravesical Implantable System
https://www.jnjlabels.com/package-insert/product-instructions-for-use/INLEXZO-ifu.pdf
Drug information
Associated long-acting platforms
bi-oval-shaped osmotic urinary catheter based delivery system
Administration route
Intravesical
Therapeutic area(s)
Use case(s)
Use of drug
Ease of administration
Frequency of administration
User acceptance
Common side effects include Frequent urination, dysuria (painful urination), and micturition urgency. INLEXZO is contraindicated in patients with a perforated bladder. Administration in such cases can lead to severe systemic exposure to Gemcitabine. Systemic exposure may cause fetal harm. Females of reproductive potential should use effective contraception during treatment and for 6 months following final removal. Males with partners of reproductive potential should use effective contraception during treatment and for 3 months following final removal.
Dosage
Available dose and strength
225 mg every 3 weeks
Maximum dose
225 mg
Recommended dosing regimen
Gemcitabine Intravesical System (225 mg of gemcitabine) into the bladder once every 3 weeks for up to 6 months (8 doses), followed by once every 12 weeks for up to 18 months (6 doses), or until persistent or recurrent NMIBC, disease progression, or unacceptable toxicity.
Additional comments
Not provided
Drug information
Drug's link(s)
Generic name
Brand name
Compound type
Drug class/category
Summary
Approval status
Regulatory authorities
Delivery device(s)
intravesical implantable device containing solid gemcitabine tablets
Scale-up and manufacturing prospects
Scale-up prospects
Projected revenue for INLEXZO is expected to reach $33 million in 2025 and increase significantly to $223 million in 2026.
Tentative equipment list for manufacturing
Tablet equipments: 1. Precision analytical balances / weigh booths 2. Powder blender / tumble mixer 3. Mini‑tablet press 4. Controlled‑humidity storage cabinets Polymer Component Fabrication Equipment: 1. Medical‑grade silicone extrusion line 2. Hydrophilic TPU film extrusion / casting machines 3. Precision die‑cutters 4. Laser micromachining system 5. Nitinol wire forming and heat‑setting furnace
Manufacturing
Process include (ISO‑classified cleanrooms required: Tablet handling/assembly: ISO 7–8; Device loading / open processes: ISO 5–7): 1. Mini‑Tablet Drug Core Production 2. Polymer Component Fabrication 3. Device Assembly 4. Sterilization & Packaging 5. In‑Vitro Release Testing (QC Lot Release)
Specific analytical instrument required for characterization of formulation
1. HPLC‑UV 2. LC‑MS 3. Durometer tester, tensile tester, microscopy, contact‑angle/swelling measurement 4. Bioburden test kit
Clinical trials
CR108889
Identifier
NCT03404791
Link
https://clinicaltrials.gov/study/NCT03404791
Phase
Phase I
Status
Completed
Sponsor
Janssen Research & Development, LLC
More details
The purpose of this study is to evaluate both the safety and tolerability of up to 4 dosing cycles of TAR-200 for 21 days per dosing cycle in the induction period.
Purpose
A Study of TAR-200 in Participants With Muscle-Invasive Urothelial Carcinoma of the Bladder Who Are Ineligible for or Refuse Cisplatin-based Chemotherapy and Who Are Unfit for Radical Cystectomy
Interventions
Intervention 1
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2017-11-20
Anticipated Date of Last Follow-up
2025-01-31
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2019-12-23
Actual Completion Date
2022-09-15
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Histological proof of non-metastatic muscle-invasive urothelial cell carcinoma of the bladder * Participant must have been as fully resected as possible per the physician's judgment * Participants must be deemed unfit for RC due to comorbid conditions with a risk of mortality * Participants must refuse or be deemed ineligible for cisplatin-based chemotherapy * Participant must refuse or not be eligible for radiotherapy Exclusion Criteria: * Other active malignancies * Presence of any bladder or urethral anatomic feature that in the opinion of the Investigator may prevent the safe placement, indwelling use, or removal of TAR-200 * Pyeloureteral tube externalized to the skin (ureteral stent or unilateral nephrostomy tube is allowed) * Evidence of bladder perforation
Health status
Study type
Interventional (clinical trial)
Enrollment
35
Allocation
Not provided
Intervention model
Single group assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
TAR-200-102
Identifier
NCT02720367
Link
https://clinicaltrials.gov/study/NCT02720367
Phase
Phase I
Status
Completed
Sponsor
Taris Biomedical LLC
More details
The purpose of this study is to determine if TAR-200, an investigational drug-delivery system is safe and tolerable in patients with recurrent low or intermediate risk non-muscle-invasive bladder cancer (NMIBC) between diagnosis and transurethral resection of bladder tumors (TURBT)
Purpose
Safety and Tolerability of TAR-200 mg in Subjects With Non-Muscle-Invasive Bladder Cancer
Interventions
Intervention 1
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2016-01-01
Anticipated Date of Last Follow-up
2020-03-09
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2018-01-01
Actual Completion Date
2020-03-01
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * A documented history of histologically-confirmed low or intermediate risk urothelial carcinoma of the bladder, excluding carcinoma in situ (pTis), pathologic stage pT1 (invasive into lamina propria) and high-Grade disease, judged not to be muscle infiltrating (pT2 or greater) and accessible for resection. * Adequate laboratory parameters. * Screening urinalysis showing no clinically significant abnormalities except those attributable to bladder cancer. * Not undergoing active treatment in last 3 months for prior or concurrent neoplastic disease and have fully recovered from treatment effects. Patients undergoing concurrent hormonal therapy treatment for prostate cancer will be allowed to enroll. Exclusion Criteria: * Exposure to BCG therapy and/or any other intrave
Health status
Study type
Interventional (clinical trial)
Enrollment
12
Allocation
Not provided
Intervention model
Sequential assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
TAR-200-104
Identifier
NCT03518320
Link
https://clinicaltrials.gov/study/NCT03518320
Phase
Phase I
Status
Terminated
Sponsor
Taris Biomedical LLC
More details
The purpose of this study is to determine if TAR-200, an investigational drug delivery system, in combination with nivolumab is safe and tolerable in patients with muscle-invasive bladder cancer (MIBC) who are scheduled for radical cystectomy (RC) during an 84-day dosing cycle induction period comprised of four consecutive 21-day dosing cycles.
Purpose
Safety and Tolerability of TAR-200 and Nivolumab in Subjects With Muscle-Invasive Bladder Cancer
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2019-01-02
Anticipated Date of Last Follow-up
2024-08-23
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2019-12-11
Actual Completion Date
2019-12-11
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: 1. Histological proof of muscle-invasive urothelial carcinoma of the bladder (stage cT2-cT3b, N0-1, M0). Subjects with mixed histology are required to have documented dominant transitional cell pattern with no more than 10% squamous differentiation and 10% glandular differentiation. Micropapillary/sarcomatoid/adenocarcinoma/plasmacytoid variants are not allowed. 2. Subjects with a total tumor size of ≤2 cm following TURBT are eligible. Subjects with a tumor or tumors totaling \>2 cm at screening must undergo a second debulking TURBT to reduce the tumor(s) to ≤2 cm to be eligible for treatment. 3. Adequate bone marrow, liver, and renal function, as documented by the following laboratory assessments conducted within 28 days prior to dosing: * Hemoglobin ≥9.0 g/dL
Health status
Study type
Interventional (clinical trial)
Enrollment
13
Allocation
Not provided
Intervention model
Single group assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
TAR-200-101
Identifier
NCT02722538
Link
https://clinicaltrials.gov/study/NCT02722538
Phase
Phase I
Status
Completed
Sponsor
Taris Biomedical LLC
More details
The purpose of this study is to determine if TAR-200, an investigational drug-delivery system, is safe and tolerable in patients with muscle-invasive bladder cancer (MIBC) between diagnosis and radical cystectomy (RC).
Purpose
Safety and Tolerability of GemRIS 225 mg in Subjects With Muscle-Invasive Bladder Cancer
Interventions
Intervention 1
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2016-05-31
Anticipated Date of Last Follow-up
2023-09-15
Estimated Primary Completion Date
Not provided
Estimated Completion Date
Not provided
Actual Primary Completion Date
2019-05-02
Actual Completion Date
2019-05-02
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Histological proof of muscle-invasive transitional cell carcinoma of the bladder (stage II-III). Subjects with evidence of metastatic nodal disease to the obuturator or presacral lymph nodes only may be included (N1 M0). Subjects with any degree of fixation of the pelvic sidewall are not eligible. * In Arm 1, subjects must have residual visible tumor following TURBT. In Arm 2, subjects must be fully resected (i.e., no visible tumor or as little tumor as possible) after restaging TURBT 2-6 weeks prior to Study Day 0. * Adequate bone marrow, liver, and renal function, as assessed by the following requirements conducted within 21 days prior to dosing: 1. Hemoglobin ≥ 9.0 g/dL 2. Absolute neutrophil count (ANC) ≥ 1,500/mm3 3. Platelet count ≥ 100,000/mm3 4. Tota
Health status
Study type
Interventional (clinical trial)
Enrollment
23
Allocation
Not provided
Intervention model
Sequential assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
SunRISe-1
Identifier
NCT04640623
Link
https://clinicaltrials.gov/study/NCT04640623
Phase
Phase II
Status
Not provided
Sponsor
Janssen Research & Development, LLC
More details
The purpose of this study is to evaluate the overall complete response (CR) rate in participants treated with TAR-200 in combination with cetrelimab (Cohort 1), or TAR-200 alone (Cohort 2), or cetrelimab alone (Cohort 3) with Carcinoma in Situ (CIS), with or without concomitant high-grade Ta or T1 papillary disease; and disease-free survival (DFS) in participants treated with TAR-200 alone with papillary disease only (Cohort 4).
Purpose
A Study of TAR-200 in Combination With Cetrelimab, TAR-200 Alone, or Cetrelimab Alone in Participants With Non-Muscle Invasive Bladder Cancer (NMIBC) Unresponsive to Intravesical Bacillus Calmette-Gué
Interventions
Intervention 1
Intervention 2
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2020-12-18
Anticipated Date of Last Follow-up
2026-01-15
Estimated Primary Completion Date
Not provided
Estimated Completion Date
2027-09-30
Actual Primary Completion Date
2025-07-03
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Histologically confirmed diagnosis of persistent or recurrent high-risk non-muscle invasive bladder cancer (HR-NMIBC), (carcinoma in situ \[CIS\] or tumor in situ \[Tis\]), with or without papillary disease (T1, high-grade Ta) or papillary disease only (high-grade Ta or any T1 and absence of CIS), within 12 months of completion of the last dose of Bacillus Calmette-Guerin (BCG) therapy, in participants who have received adequate BCG. Mixed histology tumors are allowed if urothelial differentiation (transitional cell histology) is predominant. However, the presence of neuroendocrine, micropapillary, signet ring cell, plasmacytoid, or sarcomatoid features will make a participant ineligible. For participants with lamina propria invasion (T1) on the screening biopsy/ tra
Health status
Study type
Interventional (clinical trial)
Enrollment
220
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
SunRISe-5
Identifier
NCT06211764
Link
https://clinicaltrials.gov/study/NCT06211764
Phase
Phase III
Status
Not provided
Sponsor
Janssen Research & Development, LLC
More details
The purpose of this study is to compare disease free survival (DFS) in participants with recurrence of papillary-only high-risk non-muscle-invasive bladder cancer (HR-NMIBC) within 1 year of last dose of Bacillus Calmette-Guérin (BCG) therapy and who refused or are unfit for Radical Cystectomy (RC), receiving TAR-200 versus investigator's choice of single agent intravesical chemotherapy.
Purpose
A Study of TAR-200 Versus Intravesical Chemotherapy in Participants With Recurrent High-Risk Non-Muscle-Invasive Bladder Cancer (HR-NMIBC) After Bacillus Calmette-Guérin (BCG)
Interventions
Intervention 1
Intervention 2
Intervention 3
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2024-04-09
Anticipated Date of Last Follow-up
2026-01-15
Estimated Primary Completion Date
2030-11-27
Estimated Completion Date
2031-04-14
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Histologically confirmed diagnosis by local pathology (within 90 days of documented informed consent) of recurrent, papillary-only high-risk non-muscle-invasive bladder cancer (HR-NMIBC) \[defined as high-grade Ta or any T1, no carcinoma in situ (CIS)\] * Participants with variant histologic subtypes are allowed if tumor(s) demonstrate urothelial (transitional cell histology) predominance. However, neuroendocrine, and small cell variants will be excluded * Participants must be ineligible for or have elected not to undergo Radical Cystectomy (RC) * Have an Eastern Cooperative Oncology Group (ECOG) performance status Grade of 0, 1, or 2 Exclusion Criteria: * Presence of CIS at any point from time of diagnosis of papillary-only HR-NMIBC recurrence to randomization. Ad
Health status
Study type
Interventional (clinical trial)
Enrollment
272
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
SunRISe-2
Identifier
NCT04658862
Link
https://clinicaltrials.gov/study/NCT04658862
Phase
Phase III
Status
Not provided
Sponsor
Janssen Research & Development, LLC
More details
The purpose of study is to compare bladder intact-event free survival (BI-EFS) in participants receiving TAR-200 in combination with intravenous (IV) cetrelimab versus concurrent chemoradiotherapy.
Purpose
A Study of TAR-200 in Combination With Cetrelimab Versus Concurrent Chemoradiotherapy in Participants With Muscle-invasive Bladder Cancer (MIBC) of the Bladder
Interventions
Intervention 1
Intervention 2
Intervention 3
Intervention 4
Intervention 5
Countries
Sites / Institutions
Not provided
Trials dates
Anticipated Start Date
Not provided
Actual Start Date
2020-12-07
Anticipated Date of Last Follow-up
2026-01-15
Estimated Primary Completion Date
2026-12-30
Estimated Completion Date
2028-12-31
Actual Primary Completion Date
Not provided
Actual Completion Date
Not provided
Studied populations
Age Cohort
- Adults
- Older Adults
Genders
- All
Accepts pregnant individuals
Unspecified
Accepts lactating individuals
Unspecified
Accepts healthy individuals
No
Comments about the studied populations
Inclusion Criteria: * Ineligible for or have elected not to undergo radical cystectomy * All adverse events associated with any prior surgery and/or intravesical therapy must have resolved to Common Terminology Criteria for Adverse Events (CTCAE) version 5.0 Grade less than (\<) 2 prior to randomization * Eastern Cooperative Oncology Group (ECOG) performance status Grade 0, 1, or 2 * Thyroid function tests are within the normal range per investigator assessment (or stable on hormone supplementation). Investigators may consult an endocrinologist for participant eligibility assessment in the case of equivocal or marginal test results * Adequate bone marrow, liver, and renal function: Bone marrow function (without the support of cytokines or erythropoiesis-stimulating agent in preceding two
Health status
Study type
Interventional (clinical trial)
Enrollment
518
Allocation
Randomized
Intervention model
Parallel Assignment
Intervention model description
Not provided
Masking
Open label
Masking description
Not provided
Frequency of administration
Studied LA-formulation(s)
Studied route(s) of administration
Use case
Treatment
Key resources
Excipients
Proprietary excipients used
No proprietary excipient used
Novel excipients or existing excipients at a concentration above Inactive Ingredients Database (IID) for the specified route of administration
1. Polyethylene glycol 8000 (8.0 mg) 2. Povidone K30 (13.4 mg) 3. Urea (42.6 mg) in a single intravesical implantable system
Residual solvents used
Not provided
Patent info
Description
Drug delivery device, for bladder delivery of 75% w/w gemcitabine HC1)
Brief description
Implantable drug delivery devices include a housing defining a reservoir, a first unit within the reservoir, and a second unit within the reservoir. The first unit contains a drug and the second unit contains a functional agent that facilitates release of the drug. Intravesical drug delivery devices include a housing portion containing a drug formulation and a housing portion containing an excipient, and are configured to release the drug according to a first release profile and the excipient according to a second release profile.
Representative patent
WO2015026813
Category
Device
Patent holder
WO2015026813
Exclusivity
Not provided
Expiration date
August 19, 2034
Status
Granted: AU, BR, CA, CN, EP (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LI, LT, LU, LV, MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TR), IL, IN, JP, KR, MX, NZ, RU, SG, US, VN, ZA
Description
Drug delivery device wholly deployable within the bladder
Brief description
Intravesical devices are provided that are wholly deployable within the bladder of a patient in need of treatment and are well tolerated by the patient.
Representative patent
WO2011084712
Category
Device
Patent holder
TARIS BIOMEDICAL LLC
Exclusivity
Not provided
Expiration date
December 17, 2030
Status
Granted: AU, CA, EP (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LI, LT, LU, LV, MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TR), JP, US Pending: IL
Description
Drug delivery device and drug tablet comprising a local anaesthetic agent
Brief description
A drug delivery device is provided that includes a device body, a number of solid, compressed drug tablets, and a retention frame. The device body includes a drug reservoir lumen and a retention frame lumen. The number of solid, compressed drug tablets are positioned in the drug reservoir lumen, and the retention frame is positioned in the retention frame lumen. The drug tablets may be mini-tablets aligned in the drug reservoir lumen, with an interstice formed between any two adjacent drug tablets facilitating deformation of the device body. Systems and method are also provided for loading a drug delivery device.
Representative patent
WO2010151893, WO2010151896
Category
Device
Patent holder
TARIS BIOMEDICAL LLC
Exclusivity
Not provided
Expiration date
June 28, 2030
Status
Granted: CA, CN, EP (CH, DE, FR, GB, IE, DK, FI, HU, NL, SE, ES, IT), EA (KZ, RU), IN, IL, JP, KR, MX, US Pending: AU, BR Not in force: EG, NZ, PH, SG, ZA, VN
Description
Gemcitabine compound
Brief description
This invention belongs to the field of pharmaceutical chemistry, and provides a new difluoro carbohydrate and new antiviral nucleosides prepared by coupling the new carbohydrate with appropriate bases.
Representative patent
US4808614
Category
Compound
Patent holder
Eli Lilly and Company
Exclusivity
Not provided
Expiration date
May 5, 2010
Status
Expired
Supporting material
Publications
Tyson MD, Morris D, Palou J, Rodriguez O, Mir MC, Dickstein RJ, Guerrero-Ramos F, Scarpato KR, Hafron JM, Messing EM, Cutie CJ, Maffeo JC, Raybold B, Chau A, Stromberg KA, Keegan KA. Safety, Tolerability, and Preliminary Efficacy of TAR-200 in Patients With Muscle-invasive Bladder Cancer Who Refused or Were Unfit for Curative-intent Therapy: A Phase 1 Study. J Urol. 2023 May;209(5):890-900. doi: 10.1097/JU.0000000000003195. Epub 2023 Apr 7. PMID: 37026631; PMCID: PMC12721624.
Purpose: Half of patients with muscle-invasive bladder cancer worldwide may not receive curative-intent therapy. Elderly or frail patients are most affected by this unmet need. TAR-200 is a novel, intravesical drug delivery system that provides sustained, local release of gemcitabine into the bladder over a 21-day dosing cycle. The phase 1 TAR-200-103 study evaluated the safety, tolerability, and preliminary efficacy of TAR-200 in patients with muscle-invasive bladder cancer who either refused or were unfit for curative-intent therapy.
Materials and methods: Eligible patients had cT2-cT3bN0M0 urothelial carcinoma of the bladder. TAR-200 was inserted for 4 consecutive 21-day cycles over 84 days. The primary end points were safety and tolerability at 84 days. Secondary end points included rates of clinical complete response and partial response as determined by cystoscopy, biopsy, and imaging; duration of response; and overall survival.
Results: Median age of the 35 enrolled patients was 84 years, and most were male (24/35, 68.6%). Treatment-emergent adverse events related to TAR-200 occurred in 15 patients. Two patients experienced treatment-emergent adverse events leading to removal of TAR-200. At 3 months, complete response and partial response rates were 31.4% (11/35) and 8.6% (3/35), respectively, yielding an overall response rate of 40.0% (14/35; 95% CI 23.9-57.9). Median overall survival and duration of response were 27.3 months (95% CI 10.1-not estimable) and 14 months (95% CI 10.6-22.7), respectively. Progression-free rate at 12 months was 70.5%.
Conclusions: TAR-200 was generally safe, well tolerated, and had beneficial preliminary efficacy in this elderly and frail cohort with limited treatment options.
Pradere B, Schuit M, Guerrero-Ramos F, Shariat SF, Kitamura H, Jacob JM, Bao Y, Heesakkers J, Peters KM, Cahn DJ, De Troyer B, Herrera Imbroda B, Morris DS, Pieczonka CM, Wei Q, Bhanvadia S, Somer R, Jessner W, Triantos S, Sánchez de Llano C, Maffeo JC, Sweiti H, Psutka SP. Side effect management and procedural best practices with indwelling intravesical drug-releasing systems in the treatment of bladder cancer: recommendations from expert panels. Curr Opin Urol. 2025 Oct 8;36(1):123–33. doi: 10.1097/MOU.0000000000001350. Epub ahead of print. PMID: 41065373; PMCID: PMC12700673.
Purpose of review
To provide expert recommendations for side effect management in patients with bladder cancer receiving intravesical-drug releasing system (iDRS) treatment and for optimizing iDRS insertion procedure success.
Recent findings
Indwelling iDRS are designed to provide sustained local exposure to therapy. In clinical trials, frequent side effects of iDRS treatment were lower urinary tract symptoms (LUTS) (e.g., dysuria, pollakiuria, micturition urgency), urinary tract infections (UTIs), and hematuria. These side effects are generally low grade, but if not properly managed, may lead to treatment interruptions or discontinuations. As data are limited, practical recommendations based on expert opinion for the management of common side effects and best practices for iDRS insertion procedures may improve treatment adherence and optimize outcomes in patients with bladder cancer receiving iDRS.
Summary
Two separate expert panels were convened to develop recommendations for side effect management with iDRS and optimizing iDRS insertion procedure success. Stepwise treatment-specific management strategies for LUTS, UTIs, and hematuria in patients receiving iDRS treatment that are familiar to practicing urologists are presented, including considerations for continuation or discontinuation of iDRS treatment. Several advanced techniques can be considered to improve iDRS insertions based on variations in patient anatomy.
Daneshmand, S., Brummelhuis, I. S. G., Pohar, K. S., Steinberg, G. D., Aron, M., Cutie, C. J., Keegan, K. A., Maffeo, J. C., Reynolds, D. L., Raybold, B., Chau, A., & Witjes, J. A. (2022). The safety, tolerability, and efficacy of a neoadjuvant gemcitabine intravesical drug delivery system (TAR-200) in muscle-invasive bladder cancer patients: a phase I trial. Urologic oncology, 40(7), 344.e1–344.e9. https://doi.org/10.1016/j.urolonc.2022.02.009
Objectives: Neoadjuvant chemotherapy and radical cystectomy (RC) are underutilized standards of care for the treatment of muscle-invasive bladder cancer (MIBC) due to high patient burden from systemic toxicities and postoperative complications, respectively. TAR-200 is a novel intravesical drug delivery system developed to release gemcitabine into the bladder urine continuously, resulting in distribution of drug into stromal layers of the bladder. The primary aim of the TAR-200-101 study was to evaluate the safety of TAR-200 in patients with MIBC prior to RC (NCT02722538).
Methods and materials: This phase I, open-label study was conducted across 6 US and European sites. Eligible patients were aged ≥18 years with histologically confirmed T2a-T3b N0-N1 M0 urothelial cancer and had refusal or were ineligible to receive cisplatin-based combination chemotherapy. Two arms were enrolled serially. Patients in Arm 1 had residual tumor >3 cm after transurethral resection of bladder tumor (TURBT); those in Arm 2 had undergone maximal TURBT (residual tumor <3 cm). Patients received two 7-day cycles of intravesical gemcitabine delivery via TAR-200 before undergoing RC. Primary outcome was safety; secondary outcomes were tolerability, pharmacokinetics, and preliminary efficacy.
Results: Of 23 patients in the intention-to-treat population (11 in Arm 1, 12 in Arm 2), 20 completed both dosing cycles of TAR-200. No patients were classified as intolerant to TAR-200. Ten patients (4 in Arm 1, 6 in Arm 2) experienced ≥1 treatment-emergent adverse events (TEAEs). The most common TAR-200-related TEAEs were pollakiuria (n = 3) and urinary incontinence (n = 2). All TEAEs prior to RC were grade ≤2; 1 patient in Arm 2 experienced a grade 3 non-treatment-related TEAE. Plasma gemcitabine levels were undetectable. In Arm 1, those with residual tumor, 4 of 10 patients exhibited pathologic downstaging; 1 experienced a complete response (CR) and 3 a partial response (PR). In Arm 2, those undergoing maximal TURBT, 6 of 10 patients exhibited downstaging; 3 experienced a CR and 3 a PR.
Conclusion: Controlled intravesical gemcitabine release via TAR-200 was safe and well tolerated in patients with MIBC.
Additional documents
No documents were uploaded
Useful links
Access principles

|
Collaborate for developmentConsider on a case by case basis, collaborating on developing long acting products with potential significant public health impact, especially for low- and middle-income countries (LMICs), utilising the referred to long-acting technology Not provided |

|
Share technical information for match-making assessmentProvide necessary technical information to a potential partner, under confidentiality agreement, to enable preliminary assessment of whether specific medicines of public health importance in LMICs might be compatible with the referred to long-acting technology to achieve a public health benefit Not provided |

|
Work with MPP to expand access in LMICsIn the event that a product using the referred to long-acting technology is successfully developed, the technology IP holder(s) will work with the Medicines Patent Pool towards putting in place the most appropriate strategy for timely and affordable access in low and middle-income countries, including through licensing Not provided |
Comment & Information
Not provided